正文
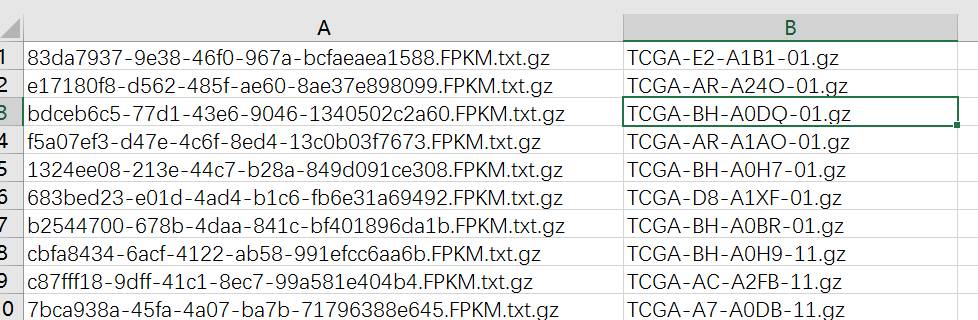
将第二列复制,并且替换-01.gz为空
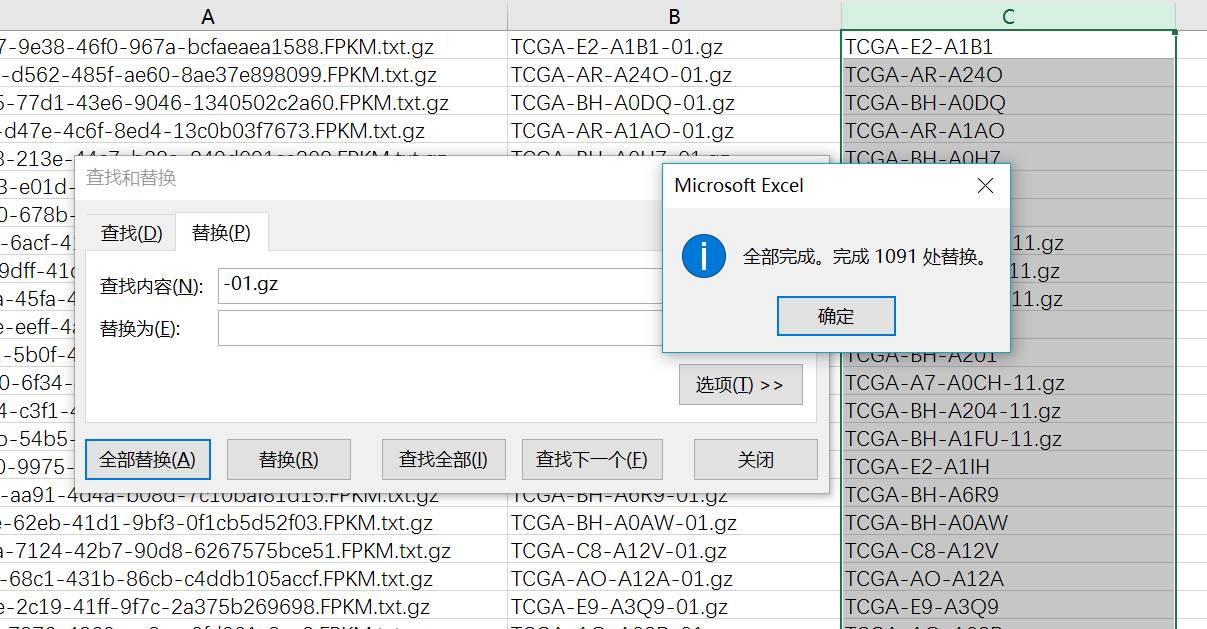
使用Excel的vlookup命令将临床病理资料的那100个样本进行映射
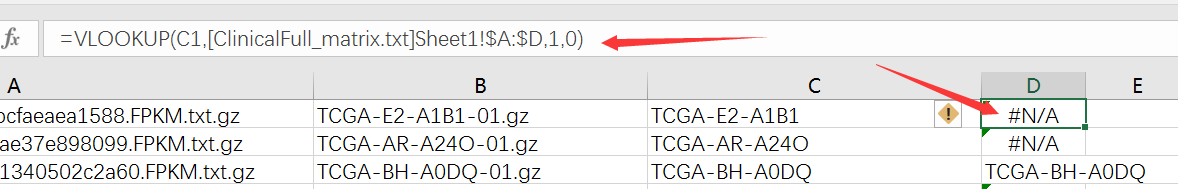
然后筛选非N/A的就得到了这一百个样本对于的RNA-seq数据信息
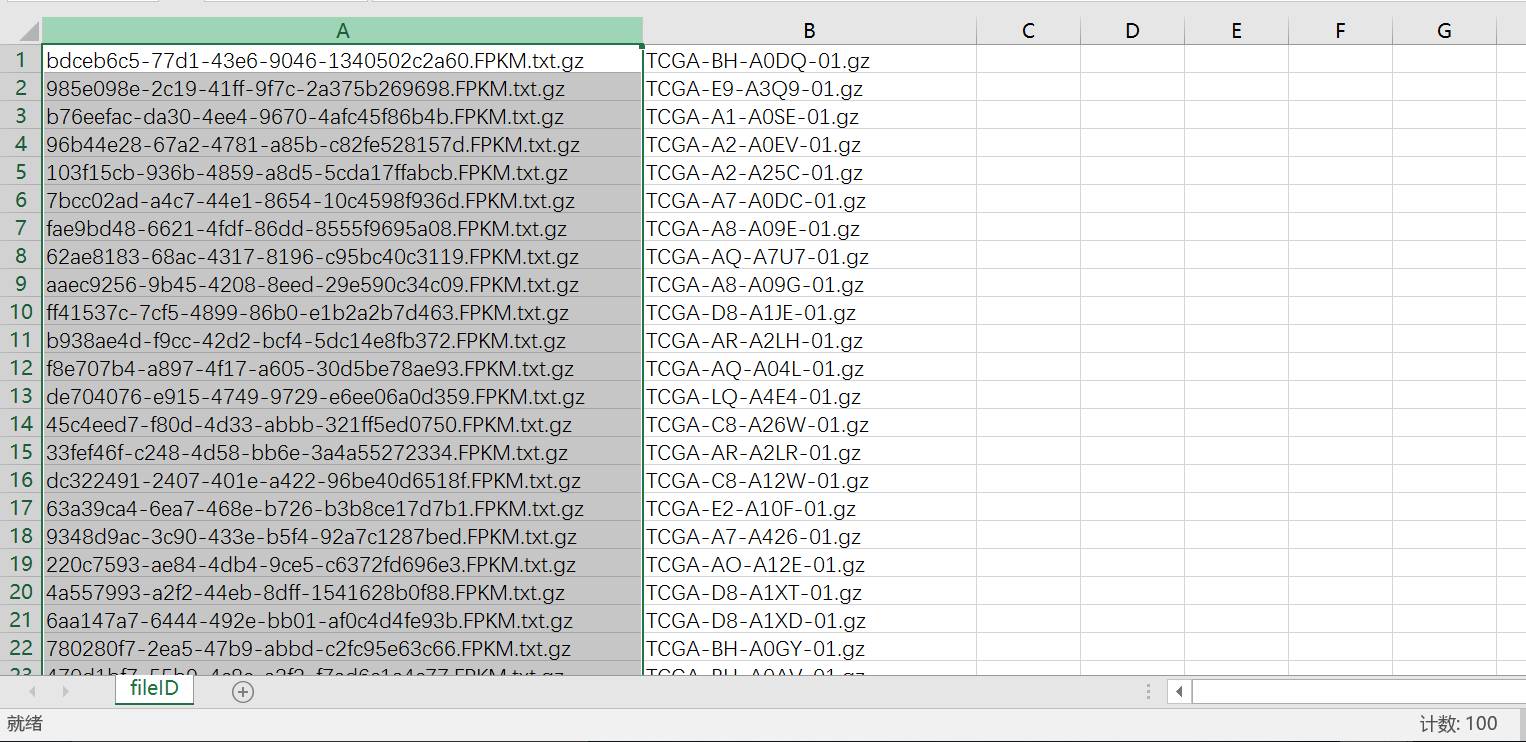
 进一步删除其他的样本,还原成fileID.tmp格式保存退出:
进一步删除其他的样本,还原成fileID.tmp格式保存退出:
然后使用TCGA简易小工具“合并文件”按钮就得到表达矩阵了,进一步使用ENSD_ID转换按钮就得到了基因表达矩阵和lncRNA表达矩阵了
#################R代码实现WGCNA##############
setwd('E:/rawData/TCGA_DATA/TCGA-BRCA')
samples=read.csv('ClinicalFull_matrix.txt',sep = '\t',row.names = 1)
dim(samples)
#[1] 100 3
expro=read.csv('Merge_matrix.txt.cv.txt',sep = '\t',row.names = 1)
dim(expro)
#[1] 24991 100
数据读取完成,从上述结果可以看出100个样本,有24991个基因,这么多基因全部用来做WGCNA很显然没有必要,我们只要选择一些具有代表性的基因就够了,这里我们采取的方式是选择在100个样本中方差较大的那些基因(意味着在不同样本中变化较大)
继续命令:
m.vars=apply(expro,1,var)
expro.upper=expro[which(m.vars>quantile(m.vars, probs = seq(0, 1, 0.25))[4]),]##选择方差最大的前25%个基因作为后续WGCNA的输入数据集
通过上述步骤拿到了6248个基因的表达谱作为WGCNA的输入数据集,进一步的我们需要看看样本之间的差异情况




